
Developing standard operating procedures and workflows for common bioinformatics data analysis tasks
Tools & Services
Bioinformatics Workflows
H3ABioNet developed standard operating procedures (SOPs) for common bioinformatics data analysis tasks. Some SOPs contain a software workflow as well as corresponding containers with all the necessary software dependencies to run the workflow. The list of SOPs and their corresponding workflows can be accessed below:
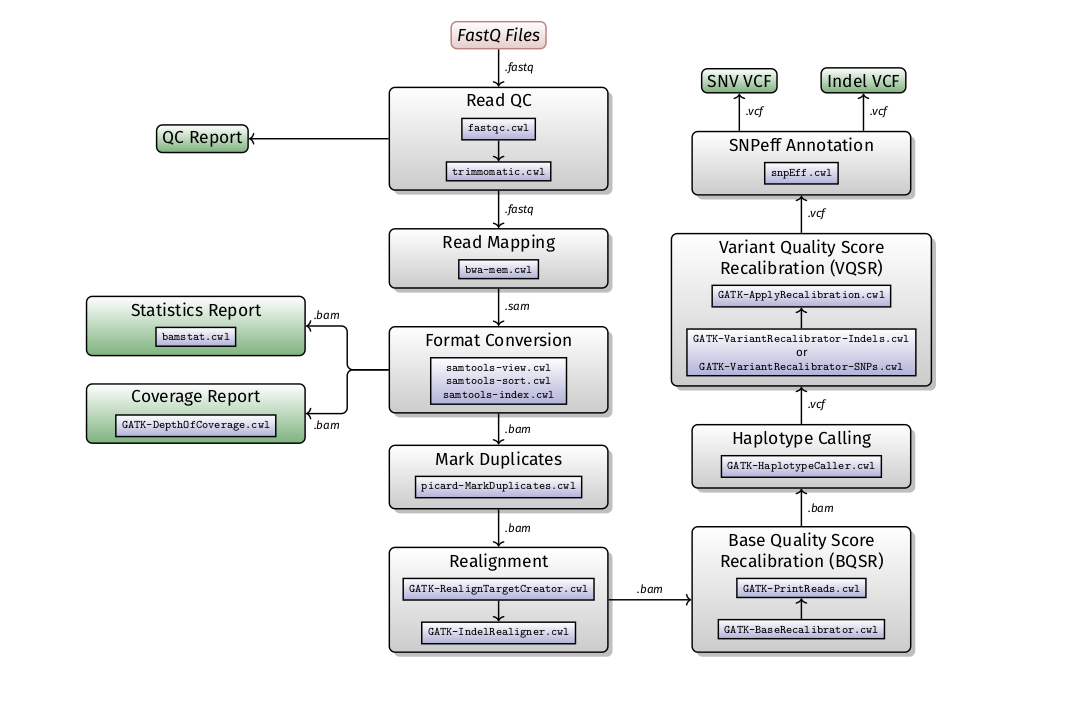
Human variant calling
A SOP for human variant calling has been developed to serves as a guideline for read QC, alignment and variant calling from exome sequencing data. We have also implement an extended version the Broad Institute’s Genome Analysis ToolKit (GATK) Best Practices version 3.5 for human variant calling (exome and genome processing). The GATK best practices for variant calling has been extensively validated and accepted as an industry standard for human NGS data analysis. Additional steps included in our pipeline include read trimmimng, quality control of reads and alignments, coverage statistics and variant annotation.
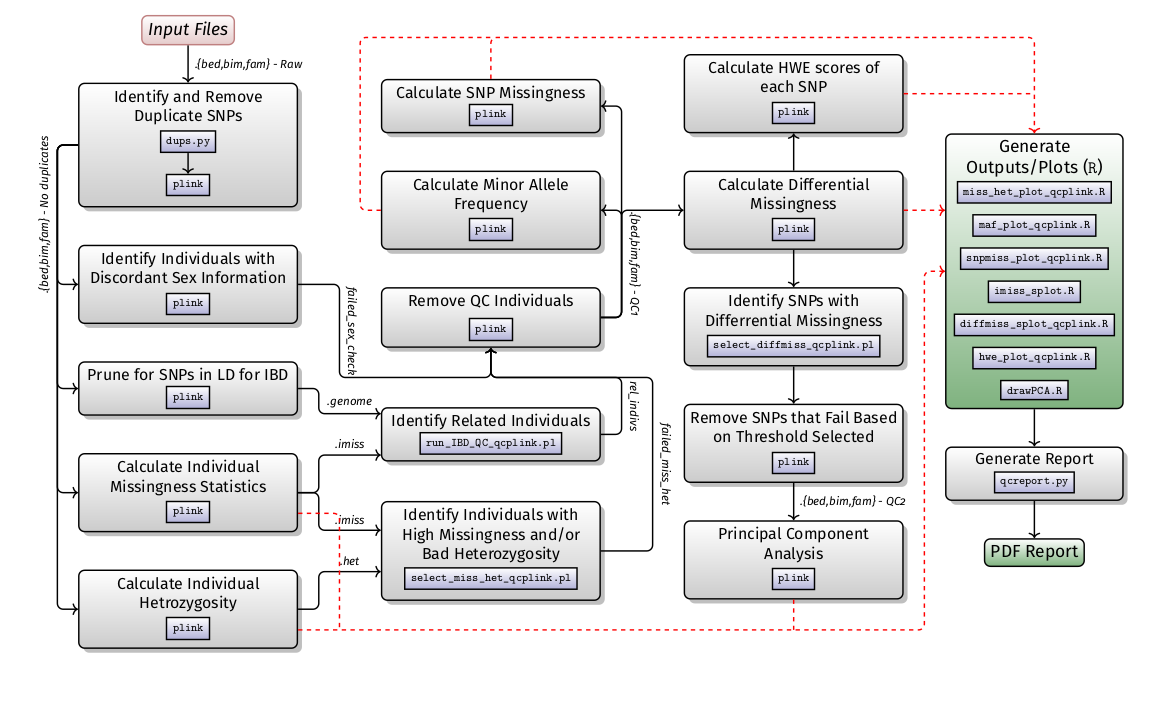
GWAS
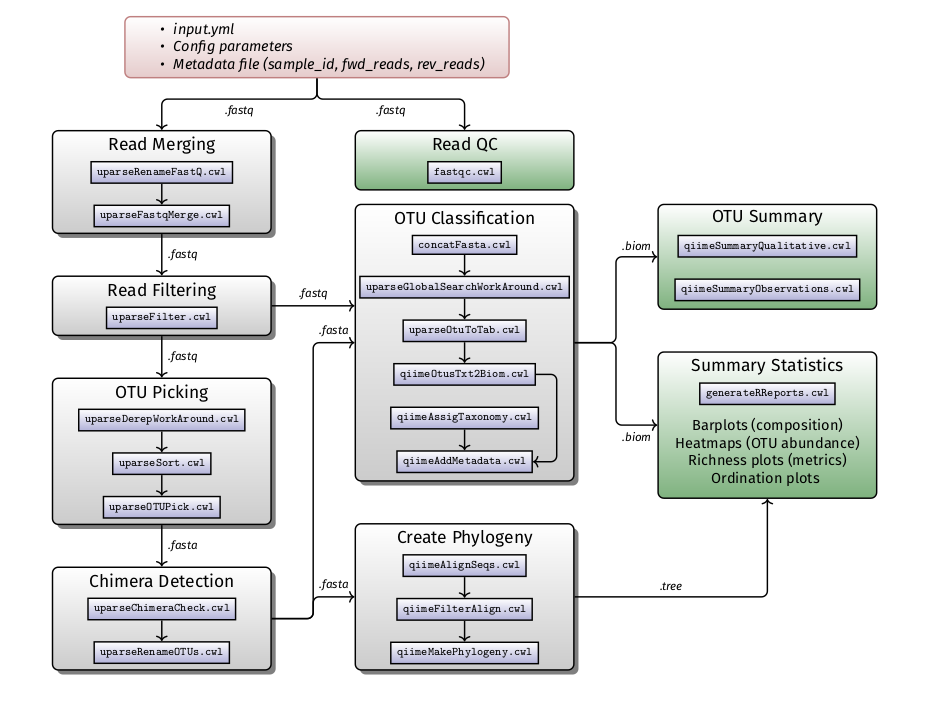
16S rDNA diversity analysis
RNA-Seq
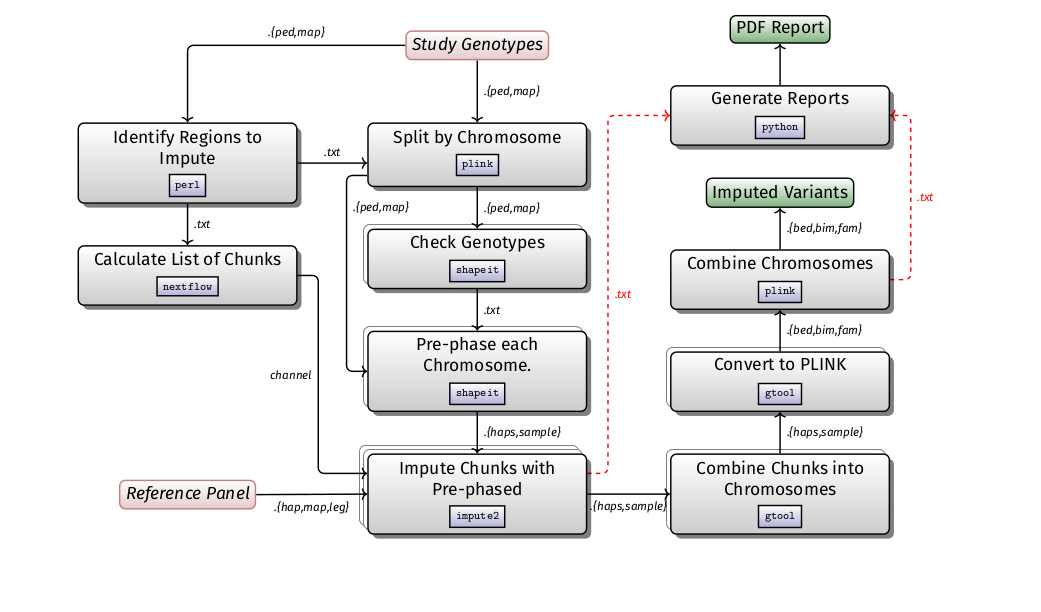
Variant discovery
Variant prioritisation