
An insight into genome-wide nucleosome positions is required to understand the local regulation of genome function. Bioinformatics tools to analyse nucleosome positions in genomes are limited. This paucity is addressed with NUCPOS, a suite that provides several utilities to analyse important aspects of nucleosomal organisation, including nucleosome density, the positioning strength of individual nucleosomes, the contribution of sequence to observed positions, and the average nucleosomal organization of specified genomic positions, such as pol II transcription start sites.
NUCPOS is available under the GNU public license (GPL-3). The C++ 11 source code may be downloaded from https://sourceforge.net/projects/nucpos/ and compiled with GCC (gcc.gnu.org) g++ version 4.7.3 or later.
Nucfrag: takes the SAM format (samtools.github.io/hts-specs/SAMv1.pdf) output file without headers generated by Bowtie2 (bowtie-bio.sourceforge.net) as input, and generates a series of data files of the number of nucleosomes centred at each base pair position of each chromosome. Nucfrag allows the selection of lower and upper fragment sizes to select subpopulations of nucleosomes, possibly with or without associated linker histone H1, from the bowtie2 output file. Additional outputs include a file of the distribution of the aligned fragment sizes in the selected size range, and data files of virtual footprints, simulating genomic areas that would be protected from nuclease cleavage by nucleosomes. The nucleosome position data files can be uploaded and viewed in a genome browser after addition of the appropriate BED or bigBed format headers (genome.ucsc.edu).
Dyad_bins: is a program that takes the nucleosome position data files generated by Nucfrag as input, and performs a binning analysis, i.e., it counts the number of times that a specific number of nucleosomes are co-aligned in the genome. The output can simply be visualized with a graphing program such as Gnuplot (www.gnuplot.info). The co-alignment of many nucleosomes at a specific genomic position generally indicates a strongly positioned nucleosome, which may have functional relevance. This analysis provides insight into the general nucleosome density and number of well-positioned nucleosomes in specified genomic regions (Fig. 1 A).
Align_dyads: is a program that takes a text file with a list of genomic positions and the nucleosome position data files generated by Nucfrag as input, and generates a data file of superimposed nucleosome positions aligned at the specified genomic positions. The user can select the number of nucleotides to include upstream and downstream of the listed positions. The program is especially useful to gain insight into the average nucleosomal organization of transcription start sites, defined replication origins, and similar functional elements in a genome. An output file is written than can be visualized in Gnuplot (Fig. 1 B).
The precise rotational and translational position of a nucleosome is determined by the DNA sequence accommodated by the nucleosome, as well as steric influences due to other proteins bound to the DNA. The contribution of intrinsic sequence effects is often useful to understand whether specific, functionally significant, well-positioned nucleosomes are precisely placed due to the inherent DNA sequence, or due other features of the genome. The utility hp_fft allows the user to quantitatively access the contribution of dinucleotide periodicities to the anisotropic flexibility of nucleosomes positioned at identified genomic positions (Fig. 1 C).
hp_fft: performs the fast Fourier transform of the distribution of each of the 16 possible dinucleotides in a sliding 128 nt window, and provides the Fourier magnitude of the distribution of each dinucleotide at a periodicity of approximately 10 nt. hp_fft takes as input the fractional occurrence of each dinucleotide at each sequence position in the sequence of interest. The factional distribution is generated with the utility dinucleotide_frequencies, which takes as input the FastA format sequence file, which may contain multiple sequences representing nucleosomes positioned at specific genomic features. hp_fft requires the open source FFTW library (www.fftw.org).
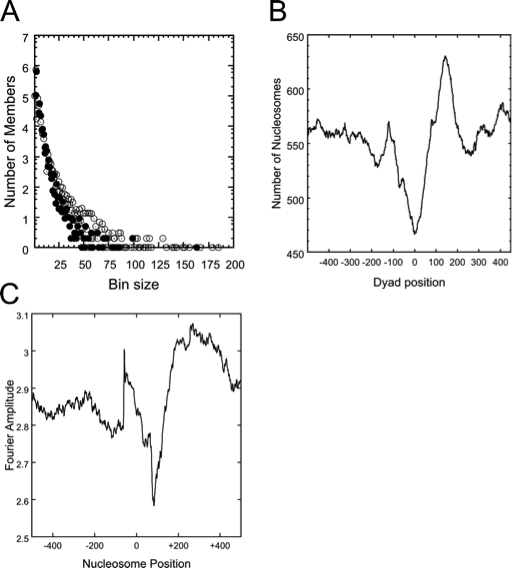
Fig. 1. (A) Distribution of co-aligned nucleosomes into bins. Nucleosomes present on coding regions (filled circles) and on non-coding regions (white circles) are shown. (B) The average nucleosomal occupancy of a polyadenylation site. Note the nucleosome depleted region at position 0, and the strongly positioned nucleosome at position 150. (C) Fourier amplitude of a genomic region. Dinucleotide distributions that could support well-positioned nucleosomes are present at positions 50 and the region 200-400. All images were generated from NUCPOS output files with Gnuplot.